Мукополисахаридозы – путь к диагнозу
Мукополисахаридозы (МПС) – это группа редких наследственных заболеваний, которые обусловлены дефицитом определенных лизосомных ферментов, участвующих в разрушении гликозаминогликанов (ГАГ), и характеризуются накоплением последних в различных органах и тканях. У части больных наблюдается медленное прогрессирование МПС, а типичные фенотипические признаки появляются в подростковом или зрелом возрасте, что в значительной степени затрудняется диагностику. Одним из типичных симптомов МПС является нарастающая тугоподвижность суставах, поэтому такие больные могут обращаться за помощью к ревматологам. Особенность поражения опорно-двигательного аппарата при МПС – отсутствие локальных и системных признаков воспаления. Важное диагностическое значение имеют системные проявления, такие как пупочная и паховая грыжа, изменение черт лица, помутнение роговицы, низкий рост, увеличение печени и селезенки, рецидивирующие инфекции дыхательных путей и средний отит и др. Для подтверждения диагноза определяют экскрецию ГАГ с мочой и активность лизосомных ферментов, а также проводят молекулярно-генетическое исследование.
Метки статьи
Мукополисахаридозы (МПС) – это группа редких лизосомных болезней накопления, характеризующихся нарушением обмена гликозаминогликанов (ГАГ). Причиной каждого МПС является генетически обусловленный дефицит определенного лизосомного фермента, участвующего в разрушении ГАГ. Практически все МПС (за исключением МПС II) наследуются по аутосомно-рецессивному типу и с равной частотой встречаются у мальчиков и девочек. МПС II – это Х-сцепленное рецессивное заболевание, которое развивается у мальчиков, хотя описаны отдельные случаи и у девочек [1].
Накопление ГАГ в лизосомах различных тканей сопровождается разнообразными системными проявлениями, в том числе поражением опорно-двигательного аппарата, сердца, нервной системы, органа зрения и др., и приводит к прогрессирующему ухудшению функции внутренних органов. МПС в целом характеризуются тяжелым течением и неблагоприятным прогнозом, поэтому многие пациенты умирают в детском или подростковом возрасте. Однако возможно и более легкое течение заболевания, в частности МПС I и МПС VI, когда симптомы появляются в подростковом или старшем возрасте и нарастают более постепенно, а пациенты доживают до зрелого возраста [2,3]. В таких случаях диагноз нередко устанавливают с опозданием, а МПС длительно протекает под маской других болезней, прежде всего ревматических. Выделение легкого варианта течения МПС весьма условно, так как при медленном прогрессировании заболевания в конечном итоге развивается тяжелое поражение отдельных органов, которое приводит к инвалидизации пациентов и может потребовать оперативного вмешательства (например, протезирование тазобедренного сустава, имплантация искусственного клапана сердца, декомпрессия спинного мозга) [4]. МПС – это неоднородная группа заболеваний, которые имеют как общие фенотипические признаки, так и существенные различия (табл. 1, 2)
| • МПС I, II и VII – системные заболевания, поражающие различные органы и ткани, включая ЦНС; неврологические нарушения не бывают изолированными. |
| • МПС III характеризуется поражением ЦНС при отсутствии соматических проявлений. |
| • МПС IV поражает в основном опорно-двигательный аппарат и не сопровождается снижением интеллекта. |
| • При МПС VI наблюдается поражение различных органов и систем, интеллект остается нормальным. |
| Тип | Название | Фермент | Ген | Тип наследования | Лечение |
|---|---|---|---|---|---|
| МПС I | Синдромы Гурлера, Шейе или Гурлера-Шейе | α-L-идуронидаза | IDUA 4p16.3 | Аутосомно-рецессивный | Ларонидаза |
| МПС II | Синдром Хантера | Идуронат-2-сульфатаза | IDS Xq28 | X-сцепленный рецессивный | Идурсульфаза |
| МПС IIIA | Синдром Санфилиппо A | Гепаран-N-сульфатаза | SGSH 17q25.3 | Аутосомно-рецессивный | Разрабатывается |
| МПС IIIB | Синдром Санфилиппо В | α-N-ацетилглюкозаминидаза | NAGLU 17q21 | Аутосомно-рецессивный | |
| МПС IIIC | Синдром Санфилиппо С | Ацетил-КоА α-глюкозамин-ацетил-трансфераза | HGSNAT 8p11.1 | Аутосомно-рецессивный | |
| МПС IIID | Синдром Санфилиппо D | N-ацетилглюкозамин-6-сульфатаза | GNS 12q14 | Аутосомно-рецессивный | — |
| MПС IVA | Синдром Моркио А | Галактозамин-6 сульфатсульфатаза | GALNS 16q24.3 | Аутосомно-рецессивный | Элосульфаза |
| MПС IVB | Синдром Моркио В | β-Галактозидаза | GLB1 3p21.33 | Аутосомно-рецессивный | — |
| MПС VI | Синдром Марото-Лами | Арилсульфатаза B | ARSB 5q11.q13 | Аутосомно-рецессивный | Галсульфаза |
| МПС VII | Синдром Слая | β-Глюкуронидаза | GUSB 7q21.11 | Аутосомно-рецессивный | Разрабатыва |
| МПС IX | Синдром Натовича | Гиалуронидаза I | AH 3p21.3-p21.2 | Аутосомно-рецессивный | — |
Своевременная диагностика МПС сегодня приобрела особое значение, учитывая возможность заместительной терапии рекомбинантными ферментами, такими как идурсульфаза (МПС II), ларонидаза (МПС I), галсульфаза (МПС VI) и элосульфаза (МПС IVA), которые позволяют улучшить состояние больных или по крайней мере затормозить прогрессирование заболевания [5]. Ферментозаместительная терапия (ФЗТ) более эффективна, если ее начинают на более раннем этапе, когда еще отсутствуют необратимые проявления болезни.
Трудности диагностики МПС
МПС относятся к очень редким (орфанным) заболеваниям. В разных странах различные МПС регистрировали с частотой 1 на 16000-29000 живых новорожденных [6,7], а в 2007 году в Скандинавских странах распространенность МПС составила всего 4-7 случаев на 1 млн населения [8]. В связи с этим информированность врачей, особенно наблюдающих взрослых пациентов, о МПС низкая. Дополнительные сложности в диагностике возникают при более легком течении МПС, особенно при отсутствии типичных фенотипических проявлений, таких как низкий рост и характерные черты лица. Например, в зависимости от клинических проявлений и течения выделяют три формы МПС I – тяжелую (синдром Гурлера), промежуточную (синдром Гурлера-Шейе) и более легкую (синдром Шейе). Во всех случаях причиной заболевания является мутация гена, кодирующего α-L-идуронидазу. У пациентов с синдромом Гурлера симптомы появляются в раннем детском возрасте и часто наблюдается тяжелое поражение ЦНС, в то время как при синдроме Шейе симптомы менее выражены и возникают значительно позднее, а когнитивные расстройства обычно отсутствуют [9]. Два варианта течения заболевания – тяжелый и более легкий – возможны и при МПС VI (синдроме Марото–Лами), обусловленном мутациями гена, кодирующего арилсульфатазу В.
В клинике им. Е.М. Тареева за последние 3 года были обследованы 5 взрослых пациентов в возрасте от 20 до 33 лет с МПС VI. У трех из них диагноз был установлен в подростковом возрасте (от 7 до 16 лет), а у двух – в возрасте 23 и 30 лет, соответственно. Необ хо димо подчеркнуть, что хотя у двух последних пациенток наблюдалось замедленное прогрессирование заболевания, тем не менее, в обоих случаях на момент госпитализации в клинику имелось тяжелое поражение опорно-двигательного аппарата с резким ограничением подвижности в суставах, пороки клапанов сердца, стеноз шейного отдела позвоночника, нарушение проходимости дыхательных путей, поражение органа зрения и др. Обе пациентки были низкого роста (132 и 146 см) [4].
Замедленное прогрессирование течение иногда на блюдается и при МПС II (синдроме Хантера). Два года назад в нашу клинику был госпитализирован 42-летний пациент с МПС II, который был диагностирован в возрасте 13 лет на основании характерных изменений внешнего вида и наличия синдрома Хантера у старшего брата и подтвержден при энзимологическом (дефицит активности идуронат-2-сульфатазы) и молекулярногенетическом (мутация с.236С>А гена IDS в гемизиготном состоянии) исследованиях [10]. В течение длительного времени состояние пациента оставалось удовлетворительным. Успешно закончил школу, а затем институт. Работал инженером на заводе. С 30-летнего возраста прогрессирующее снижение чувствительности и боли в кистях и стопах, ухудшение зрения и выпадение центральных полей зрения, однако продолжал работать. Резкое ухудшение состояния, связанное с развитием сердечной недостаточности на фоне тяжелого порока аортального клапана, было отмечено только за год до госпитализации, т.е. в возрасте около 40 лет.
При обращении к ревматологу на МПС может указывать поражение суставов, не сопровождающееся признаками воспаления, такими как припухание, повышение СОЭ и/или уровня С-реактивного белка [12]. T. Rocha Siqueira и соавт. измеряли экскрецию ГАГ с мочой у 55 пациентов в возрасте от 3 до 21 года (в среднем 9 лет) с невоспалительной артропатией неясного генеза. У всех больных определялись дискомфорт или боль в суставах, а у 2/3 – скованность [12]. Экскре ция ГАГ была повышена у 1 из 55 больных. При дополнительном обследовании у 15-летней пациентки был установлен диагноз МПС II. Хотя очевидным ограничением этого исследования было небольшое число обследованных пациентов, тем не менее, полученные данные указывают на возможную роль скрининга в диагностике более легких форм МПС.
Как заподозрить МПС?
В настоящее время известно 11 лизосомных ферментов, дефицит которых приводит к развитию 7 типов МПС [13]. Замедленное прогрессирование заболевания и более поздняя диагностика чаще отмечаются у пациентов с МПС I, IV, VI и VII, в то время как другие типы МПС обычно характеризуются тяжелым течением и более короткой продолжительностью жизни. Следует отметить, что в задачи практического врача не входит дифференциальная диагностика различных МПС – вполне достаточно заподозрить этот диагноз и направить пациента на консультацию к генетику и/или провести скрининговое исследование (определение экскреции ГАГ с мочой).
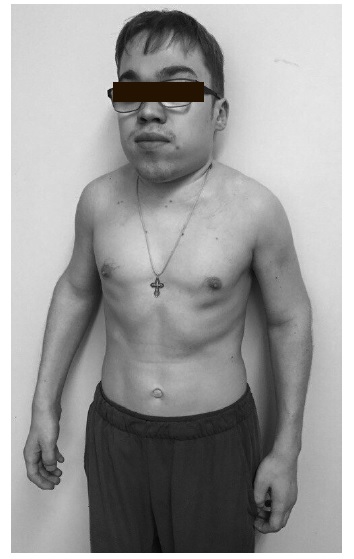
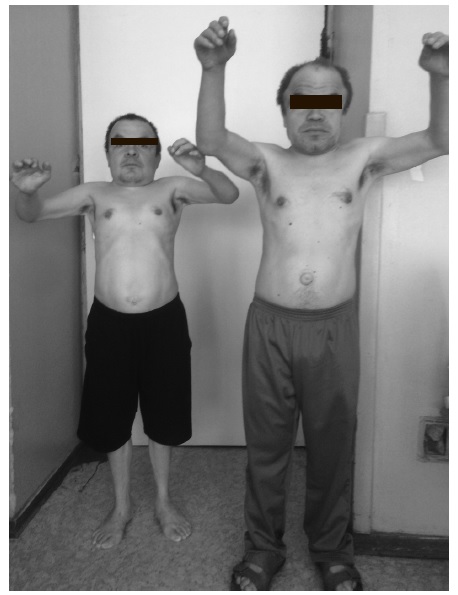
Хотя МПС представляют собой неоднородную группу болезней и отличаются по тяжести течения и частоте поражения центральной нервной системы, тем не менее, в целом клинические проявления некоторых из них достаточно однотипны и позволяют предположить наличие заболевания, особенно у пациентов старшего возраста при наличии типичного фенотипа. При осмотре пациентов с МПС прежде всего обращают на себя внимание низкий рост, непропорциональное строение скелета (короткие туловище и шея, длинные конечности), а также грубые черты лица, толстые губы, увеличение языка, запавшее переносье, увеличение расстояния между глазами (гипертелоризм) (рис. 1, 2). При тяжелом течении МПС рост пациентов не превышает 95-100 см, хотя при медленном развитии заболевания может достигать 140-150 см. Например, в нашей серии наблюдений рост 5 взрослых пациентов с МПС VI варьировался от 132 до 153 см, а рост 42-летнего пациента с МПС II составлял 158 см. В крупном исследовании среди 121 пациента с МПС VI доля взрослых составляла около 25% [14]. Средний рост больных в возрасте 19-24 и 25-56 лет равнялся 142,7±20,1 и 157,0±8,5 см, соответственно. Таким образом, по крайней мере у части больных МПС рост может быть фактически нормальным.
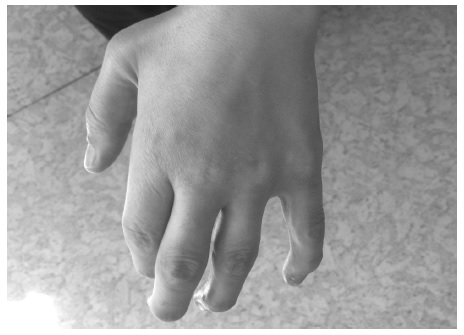
При всех МПС развивается тяжелое поражение опорно-двигательного аппарата (множественный дизостоз), которое проявляется тугоподвижностью и контрактурами суставов (в большей степени ухудшается разгибание), деформацией кистей (“когтистая лапа”) (рис. 3) и позвоночника (кифоз, сколиоз), воронкообразной грудной клеткой. Наблю даются недоразвитие таза, дисплазия головок бедренных костей и вальгусное положение шейки бедренной кости. Ограничение подвижности суставов отмечается уже в детском или подростковом возрасте, постепенно нарастает и в конечном итоге служит причиной инвалидизации больных.
Для МПС IV (синдрома Моркио), в отличие от других типов МПС, типично развитие гипермобильности суставов, обусловленной деформацией метафизов, гипоплазией костей и деградацией соединительной ткани, окружающей суставы [15].
У пациентов с МПС часто наблюдаются обструкция глотки, верхних и нижних дыхательных путей, связанная с увеличением языка и миндалин, сужением трахеи, утолщением надгортанника и голосовых связок, отложением ГАГ в слизистой оболочке бронхов. Обструкция дыхательных путей сопровождается затрудненным дыханием и громким храпом с эпизодами апноэ во время сна. Характерно развитие рецидивирующего среднего отита, вызывающего прогрессирующую тухоугость, которая обусловлена как кондуктивными, так и нейросенсорными механизмами. Причинами нарушения функции дыхания могут быть также небольшие размеры и малоподвижность грудной клетки, растя жение живота в сочетании с кифозом, сколиозом и значительным поясничным лордозом, а также рецидивирующие инфекции нижних дыхательных путей.
Еще одно типичное проявление МПС – поражение клапанов сердца, частота которого достигает 60-90%. С. Wippermann и соавт. обследовали 84 больных в возрасте от 1 до 47 лет с различными типами МПС [16]. Частота недостаточности митрального и/или аортального клапана составила 75,0%, однако тяжелая митральная или аортальная регургитация наблюдалась только в 4,8% и 8,3% случаев, соответственно. Частота пороков клапанов сердца достигала 89-100% у больных МПС I, II и VI, но была ниже у пациентов с МПС III и IV – 3366%. В другом исследовании у 28 больных МПС VI частота поражения митрального клапана составила 96%, трикуспидального – 71% и аортального – 43% [17]. Следует отметить, что, в отличие от некоторых других лизосомных болезней накопления, таких как болезнь Фабри, для МПС не характерно тяжелое поражение миокарда.
У большинства больных МПС I, VI и VII часто отмечается помутнение роговицы, в то время как при других типах МПС оно отсутствует [18].
У пациентов с тяжелыми формами МПС I и II наб лю дается поражение ЦНС (поведенческие расстрои ̆ства, задержка умственного развития, ухудшение интеллекта, тяжелая когнитивная дисфункция) [10]. Выраженные неврологические и когнитивные расстройства характерны также для МПС III. В то же время у большинства пациентов с МПС VI сохраняется нормальный интеллект.
МПС I, II и VI могут привести к развитию синдрома запястного канала, проявляющегося стойкой болью и онемением пальцев кисти в результате сдавления срединного нерва между костями и сухожилиями мышц запястья. Возможно также сдавление спинного мозга вследствие сужения спинно-мозгового канала и нестабильности атлантоаксиального канала. Компрессион ная миелопатия может осложниться слабостью в нижних конечностях и спастической параплегией или квадриплегией.
Диагноз и дифференциальный диагноз
Алгоритм диагностики и дифференциальной диагностики МПС у пациентов с поражением опорно-двигательного аппарата представлен на рис. 4 [19]. Если контрактуры суставов определяются у новорожденного ребенка, то наиболее вероятен диагноз артрогрипоза – заболевания, характеризующегося врожденными контрактурами двух и более суставов несмежных областей в сочетании с мышечной гипо- или атрофией. Различают артрогрипоз с поражением верхних и/или нижних конечностей, генерализованный и дистальный варианты. Артрогрипоз – это не самостоятельная нозологическая форма, а скорее физический симптом, который может быть обусловлен различными причинами, например, ограничением движений плода во время его развития (многоводие, маловодие, пороки развития и опухоли матки, многоплодная беременность), нарушением развития мышц (вирусные инфекции), генетическими факторами и др.
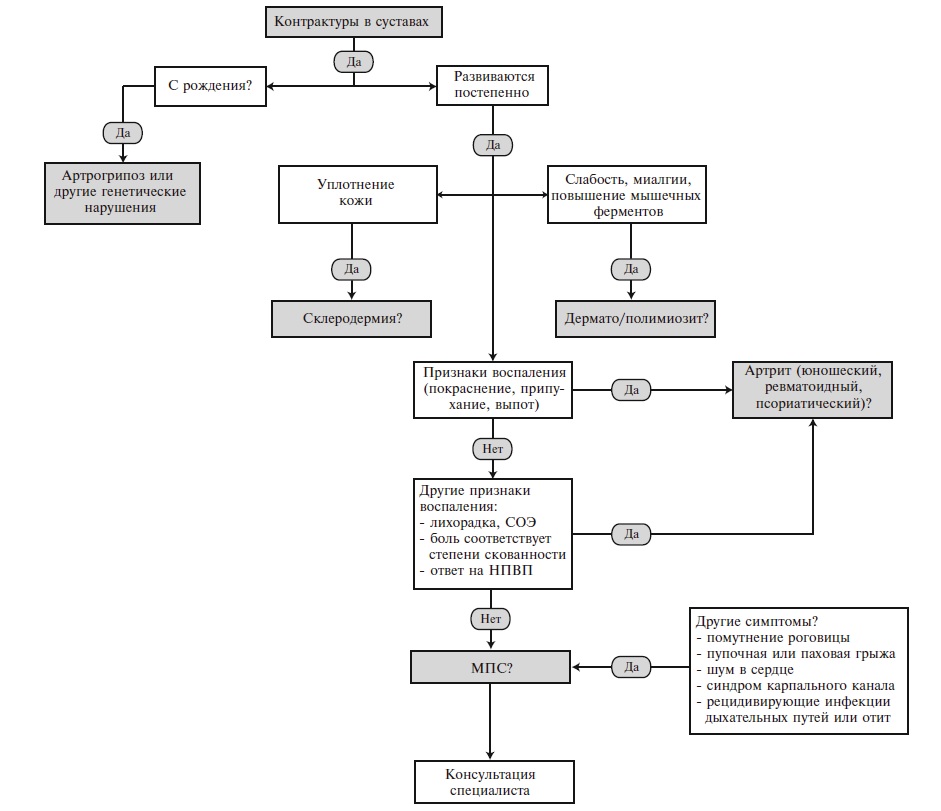
Боли и тугоподвижность суставов, появляющиеся в детском или подростковом возрасте, могут имитировать ревматические заболевания, в частности юношеский идиопатический артрит, ревматоидный или псориатический артрит. Основное значение для дифференциальной диагностики с этими заболеваниями имеют отсутствие воспалительной боли (т.е. боли, возникающей по утрам и сопровождающейся скованностью, которая уменьшается на фоне физической активности), локальных (припухание и болезненность при пальпации суставов) и системных (повышение температуры тела и/или СОЭ и уровня С-реактивного белка) признаков воспаления (рис. 4) [19]. Глюкокортикостероиды неэффективны, хотя нестероидные противовоспалительные препараты могут несколько уменьшить имеющиеся симптомы.
Признаки поражения суставов, появляющиеся в более старшем возрасте, часто расценивают как первичный остеоартроз. Дифференцировать поражение опорно-двигательного аппарата при МПС с этим забо леванием позволяют развитие артропатии в подростковом или молодом возрасте при отсутствии факторов риска первичного остеоартроза и наличие других типичных проявлений генетического заболевания (карликовый рост, измененные черты лица, порок клапана сердца, помутнение роговицы и т.п.) (табл. 3).
| Поражение опорно-двигательного аппарата |
| Контрактуры суставов, развивающиеся в раннем возрасте и не сопровождающиеся признаками воспаления или эрозивными изменениями костей |
| “Когтистая лапа” |
| Деформация позвоночника (сколиоз, кифоз, лордоз) |
| Рентгенологические признаки множественного дизостоза |
| Другие клинические проявления |
| Нарастающая “грубость” черт лица |
| Помутнение роговицы |
| Короткая ригидная шея |
| Частые респираторные инфекции, рецидивирующий средний отит, заложенность носа, шумное дыхание/храп |
| Шум в сердце |
| Пупочные и/или паховые грыжи |
| Низкий рост |
| Нарушение походки |
| Увеличение живота за счет печени и селезенки |
Скрининговым методом диагностики МПС является измерение экскреции ГАГ с мочой. Определение типа ГАГ в моче (дерматансульфат, гепарансульфат, хондороитинсульфат и кератансульфат) с помощью тонкослойной хроматографии или электрофореза имеет определенное значение для дифференциальной диагностики МПС, однако результаты этих исследований все же не позволяют установить окончательный диагноз. Экскреция ГАГ с мочой у детей, подростков и молодых людей с МПС обычно превышает таковую у здоровых людей сопоставимого возраста [13]. Однако у взрослых людей с МПС, особенно с более легкими и медленно прогрессирующими формами заболевания, она может оказаться близкой к норме. Соответственно, следует осторожно интерпретировать результаты этих тестов и продолжать обследование, если диагноз МПС представляется вероятным на основании клинических данных.
Следующий этап диагностики – определение активности лизосомных ферментов в высушенных пятнах крови, лейкоцитах или фибробластах. Анализ высушенных пятен крови обычно проводят в тех случаях, когда образец необходимо отправить в лабораторию, находящуюся в другом городе или стране. Более надежным считают исследование лейкоцитов, выделенных из цельной крови, или культивированных фибробластов.
Для подтверждения диагноза проводят молекулярногенетическое исследование, которое необходимо также для выявления носителей мутантных генов и пренатальной диагностики.
Лечение мукополисахаридозов
Для патогенетической терапии МПС применяют рекомбинантные формы ферментов, дефицит которых лежит в основе развития соответствующего заболевания, в том числе ларонидазу для лечения МПС I, идурсульфазу – МПС II, галсульфазу – МПС VI, элосульфазу альфа – МПС IVa (в Российской Федерации последний препарат не зарегистрирован). Все препараты предназначены для внутривенного введения. Их эффективность и безопасность установлены как в рандомизированных, двойных слепых, плацебо-контролируемых исследованиях, так и длительных наб людательных исследованиях, позволивших изучить отдаленные эффекы ФЗТ [20].
Эффективность и безопасность галсульфазы оценивали в рандомизированном, двойном слепом, плацебоконтролируемом, 24-недельном исследовании 3 фазы у 39 больных МПС VI [21]. Критериями эффективности были толерантность к физической нагрузке и экскреция ГАГ с мочой. Лечение галсульфазой в течение 24 недель по сравнению с плацебо привело к значительному увеличению пройденной за 12 минут дистанции (р=0,025) и скорости подъема по лестнице (р=0,053) и достоверному снижению экскреции ГАГ с мочой (p<0,001).
P. Harmatz и соавт. изучали эффективность длительной терапии галсульфазой (97-260 недель) у 56 пациентов (средний возраст 12 лет; от 5 до 29 лет) с МПС VI, которые принимали участие в трех клинических исследованиях [22]. Экскреция ГАГ с мочой достоверно снизилась после начала лечения галсульфазой, а дос тиг нутый эффект сохранялся в отдаленном периоде. К концу наблюдения средняя степень снижения экскреции ГАГ составила от 71% до 79%. У подавляющего большинства пациентов (84-89%) наблюдалось стойкое увеличение толерантности к физической нагрузке, которую оценивали на основании пройденной за 6 или 12 минут дистанции и скорости подъема по лестнице. Переносимость ФЗТ была хорошей. Доля завершенных инфузий галсульфазы составила 98%. Лечение было прекращено только у 3 пациентов (один из группы плацебо). Нежелательные явления отмечались у всех пациентов, однако чаще всего были легкими или умеренно выраженными и не связанными с исследуемым препаратом.
R. Giugliani и соавт. проанализировали результаты 10-летнего наблюдения 117 пациентов с МПС VI [23]. У 55 больных проводилась ФЗТ галсульфазой в среднем в течение 6,8±2,2 лет. Через 10 лет экскреция ГАГ с мочой снизилась в среднем на 87,9%. Средний рост увеличился на 20,4±12,4 и 16,8±6,3 см у пациентов, которые начали лечение в возрасте 4-7 лет и 8-12 лет, соответственно. У пациентов, которые завершили 6-минутную пробу, пройденная дистанция увеличилась на 65,7±100,6 м, а форсированная жизненная емкость легких (ФЖЕЛ) и объем форсированного выдоха за 1 с (ОФВ1) – на 29% и 18%, соответственно. С помощью метода Каплана-Мейера было показано, что лечение галсульфазой приводит к значительному увеличению выживаемости больных МПС VI. Сходные данные были получены с помощью модели пропорционального риска Кокса. Таким образом, результаты исследования показали, что длительная терапия галсульфазой не только улучшает рост, толерантность к физической нагрузке и показатели функции легких, но и увеличивает выживаемость больных МПС VI.
Эффективность и безопасность идурсульфазы были установлены в двойном слепом, рандомизированном, плацебо-контролируемом исследовании у 96 больных в возрасте от 5 до 31 года с МПС II [24]. Первичным критерием эффективности была комбинированная конечная точка, включавшая в себя пройденную за 6 минут дистанцию и ФЖЕЛ в процентах от должной. По этому показателю через 1 год идурсульфаза достоверно превосходила плацебо. Кроме того, ФЗТ привела к достоверному увеличению объема движений в локтевом суставе, пройденной за 6 минут дистанции и ФЖЕЛ, а также достоверному уменьшению размеров печени и селезенки. Экскреция ГАГ с мочой достоверно снизилась по сравнению с плацебо. При продолжении лечения открытым методом в течение еще 2 лет были отмечены дальнейшее увеличение ФЖЕЛ и нормализация экскреции ГАГ [25]. Значительное увеличение роста при лечении идурсульфазой было выявлено при ретроспективном анализе результатов терапии у больных, включенных в регистр Hunter Outcome Survey [26].
Эффективность и безопасность ларонидазы (0,58 мг/кг/нед) изучались в 26-недельном рандомизированном, двойном слепом, плацебо-контролируемом исследовании у 45 больных МПС I [27]. Лечение ларонидазой по сравнению с плацебо привело к достоверному увеличению медианы ФЖЕЛ (р=0,009) и пройденной за 6 минут дистанции (р=0,039), а также уменьшению размеров печени и экскреции ГАГ с мочой, а у пациентов с более тяжелой формой заболева ния – к уменьшению апноэ во время сна и увеличению объема движения в плечевых суставах. Эффективность препарата была также подтверждена при мета-анализе 4 исследований, которые показали, что препарат вызывает уменьшение экскреции ГАГ с мочой, гепатомегалии и индекса массы левого желудочка и улучшение объема движений в суставах у больных МПС I [28].
Заключение
МПС – это группа редких заболеваний, которые обычно диагностируют поздно вследствие низкой информированности врачей о лизосомных болезнях накопления. Наибольшие диагностические трудности возникают при более легких формах МПС, которые характеризуются медленным развитием соматических проявлений и стертостью типичных внешних признаков. Выделение легких, или ослабленных (attenuated), форм МПС весьма условно, так как в конечном итоге у таких больных развиваются инвалидизирующие осложения, часто требующие оперативного лечения. Одним из типичных симптомов МПС I, II и VI является нарастающая тугоподвижность в суставах, поэтому такие больные могут обращаться за помощью к ревматологам. Особенность поражения опорно-двигательного аппарата при МПС – отсутствие локальных (припухания суставов и болезненности при их пальпации) и системных (повышения температуры тела и/или СОЭ и уровня С-реактивного белка) признаков воспаления. Исключить остеоартроз позволяют молодой возраст пациента и отсутствие типичных факторов риска дегенеративных заболеваний суставов. Важное диагностическое значение имеют системные проявления, такие как пупочная и паховая грыжа, изменение черт лица, помутнение роговицы, низкий рост, увеличение печени и селезенки, рецидивирующие инфекции дыхательных путей и средний отит и др. Если заподозрен диагноз МПС, то необходимо определить экскрецию ГАГ с мочой, а также измерить активность лизосомных ферментов и провести молекулярно-генетическое исследование для подтверждения диагноза.
Используемые источники
- Tuschl K, Gal A, Paschke E et al. Mucopolysaccharidosis type II in females: case report and review of literature. Pediatr Neurol 2005;32:270-2.
- Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis 2010;5:5.
- Bruni S, Lavery C, Broomfield A. The diagnostic journey of patients with muco polysaccharidosis I: A real-world survey of patient and physician experiences. Mol Genet Metab Rep 2016;8:67-73.
- Моисеев С.В., Новиков П.И., Мешков А.Д., Фомин В.В. Муко полиса ха ридоз VI типа у взрослых. Клин фармакол тер 2017;26(1):72-9.
- Моисеев С.В., Фомин В.В. Ферментозаместительная терапия лизосомных болезней накопления. Клин фармакол тер 2016;25(1):5-6.
- Nelson J. Incidence of the mucopolysaccharidoses in Northern Ireland. Hum Genet 1997;101(3):355–8.
- Nelson J, Crowhurst J, Carey B, Greed L. Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet 2003;123A(3):310–3.
- Malm G, Lund AM, Månsson J-E, Heiberg A. Mucopolysaccharidoses in the Scandinavian countries: incidence and prevalence. Acta Paediatr Oslo Nor 2008; 97(11):1577–81.
- Vijay S, Wraith JE. Clinical presentation and follow-up of patients with the attenuated phenotype of mucopolysaccharidosis type I. Acta Paediatr 2005;94:872-7.
- Моисеев С.В., Новиков П.И., Федоров К.Е. и др. Мукополисахаридоз II типа (синдром Хантера). Клин фармакол тер 2015;24(1):76-82.
- Cimaz R, Coppa GV, Koné-Paut I, et al. Joint contractures in the absence of inflammation may indicate mucopolysaccharidosis. Pediatr Rheumatol Online J 2009;7:18.
- da Rocha Siqueira TC, de Souza CFM, Lompa P, et al. Screening for attenuated forms of mucopolysaccharidoses in patients with osteoarticular problems of unknown etiology. JIMD Rep 2016;26:99–102.
- Mitrovic S, Gouze H, Gossec L, et al. Mucopolysaccharidoses seen in adults in rheumatology. Joint Bone Spine 2017;84(6):663-70.
- Swiedler SJ, Beck M, Bajbouj M, et al. Threshold effect of urinary glycosaminoglycans and the walk test as indicators of disease progression in a survey of subjects with Mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). Am J Med Genet 2005;134A:144-50.
- Montaño AM, Tomatsu S, Gottesman GS, et al. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007;30(2):165–74.
- Wippermann CF, Beck M, Schranz D, et al. Mitral and aortic regurgitation in 84 patients with mucopolysaccharidoses. Eur J Pediatr 1995;154(2):98-101.
- Azevedo AC, Schwartz IV, Kalakun L, et al. Clinical and biochemical study of 28 patients with mucopolysaccharidosis type VI. Clin Genet 2004;66:208-13.
- Summers CG, Ashworth JL. Ocular manifestations as key features for diagnosing mucopolysaccharidoses. Rheumatology 2011;50(Suppl 5):v34-v40.
- Lehman T, Miller N, Norquist B, et al. Diagnosis of the mucopolysaccharidoses. Rheumatology 2011;50:41-8.
- Намазова-Баранова Л.С., Вашакмадзе Н.Д., Бабайкина М.А. и др. Эффек тивность современных методов лечения пациентов с мукополисахаридозом I типа. Педиатрическая фармакология 2014;11(6):76-9.
- Harmatz P, Giugliani R, Schwartz I, et al; MPS VI Phase 3 Study Group. Enzyme replacement therapy for mucopolysaccharidosis VI: a phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. J Pediatr 2006;148(4):533-9.
- Harmatz P, Giugliani R, Schwartz IV, et al; MPS VI Study Group. Long-term follow-up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: Final results of three clinical studies of recombinant human N-acetylgalactosamine 4-sulfatase. Mol Genet Metab 2008;94(4):469-75.
- Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome) — 10 year follow-up of patients who previously participated in an MPS VI survey study. Am J Med Genet 2014;164(8):1953–64.
- Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med 2006;8(8):465–73.
- Muenzer J, Beck M, Eng C, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med 2011;13(2):95-101.
- Jones SA, Parini R, Harmatz P, et al; HOS Natural History Working Group on behalf of HOS Investigators. The effect of idursulfase on growth in patients with Hunter syndrome: data from the Hunter Outcome Survey (HOS). Mol Genet Metab 2013;109(1):41-8.
- Wraith JE, Clarke LA, Beck M, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr 2004;144(5):581-8.
- Dornelles AD, Artigalás O, da Silva AA, et al. Efficacy and safety of intravenous laronidase for mucopolysaccharidosis type I: A systematic review and meta-analysis. PLoS One 2017;12(8):e0184065.
Мукополисахаридоз
Мукополисахаридоз – это наследственная патология, которая относится к лизосомным болезням накопления.
О заболевании
Лизосомные болезни накопления могут иметь очень вариабельные симптомы, что связано с поражением в той или ной степени практически всех органов и систем. Патология может впервые дать о себе знать в любом возрасте. Обычно чем тяжелее заболевание, тем раньше оно дебютирует. Лизосомные болезни накопления характеризуются прогрессирующим течением.
В структуре патологий лизосомного дефекта основное место занимают мукополисахаридозы (МПС). Для этой болезни характерна недостаточность определенных ферментных систем, которые локализованы в лизосомах внутри клетки. Эти ферменты участвуют в деградации мукополисахаридов, в частности гликозаминогликанов. Последние являются основным компонентом соединительнотканных структур. Ферментативные нарушения блокируют нормальные биохимические реакции, в результате чего в тканях накапливаются метаболиты мукополисахаридов (хондроитинсульфат В и гепаранмоносульфат). В результате этих накоплений соединительная ткань преждевременно повреждается, что и приводит к появлению разнообразной симптоматики.
Диагностируется заболевание на основании высокоспециализированных исследований. Лечение раньше проводилось только симптоматическое. Но к настоящему времени разработана также этиопатогенетическая терапия. Она может проводиться по 2 направлениям – лекарственное замещение функции поврежденного фермента или пересадка стволовых клеток крови.
Виды
Мутации могут вызывать разные дефекты ферментативных систем и тем самым определять тяжесть заболевания. На основании этого дифференцируется 14 типов заболеваний, в рамках которых выделяются дополнительные разновидности.
Наиболее распространенными являются 2 класса мукополисахаридозов:
- первый, представителем которого является синдром Хантера;
- второй, который делится на три категории: синдром Гурлера (имеет тяжелое течение), HurlerScheie синдром (характеризуется умеренной степенью прогрессирования и выраженности симптоматики) и Scheie синдром (самый легкий вариант).
Согласно современным рекомендациям, в рамках второго типа мукополисахаридоза принято выделять всего 2 разновидности – тяжелый подтип (синдром Гурлера) и ослабленный подтип (относятся 2 оставшихся синдрома).
Симптомы мукополисахаридоза
Подозрительными симптомопризнаками мукополисахаридоза в детском возрасте могут быть следующие:
- изменение визуальных характеристик лицевой области;
- скованность суставов и ограничение мобильности;
- деформации костной системы;
- большой размер головы;
- частые простудные заболевания;
- увеличение размеров сердца, выявленное с помощью УЗИ;
- неоднократно повторяющееся воспаление уха;
- грыжи различной локализации;
- переднезаднее искривление позвоночника в поясничной отделе, которое может приводить к формированию горба.
Проявления заболевания очень вариабельны, что существенно затрудняет клиническую диагностику:
- 1. Мыслительные и познавательные расстройства.
Тяжелая форма мукополисахаридоза приводит к прогрессирующему поражению центральной нервной системы. Когнитивные расстройства расцениваются как важные дифференциально-диагностические признаки. При тяжелом течении задержка умственного развития появляется еще в годовалом возрасте. Легкие формы мукополисахаридоза имеют более благоприятный прогноз, т.к. познавательная функция может быть нормальной или только с небольшим дефицитом. Поведение детей при мукополисахаридозе первого типа отличается определенной заторможенностью, в противоположность этому при втором и третьем подтипе заболевания дети проявляют агрессию и гиперактивность.
- 2. Неврологические нарушения.
Для тяжелых форм мукополисахаридоза характерно развитие гидроцефалии, в то время как при легких вариантах – она отсутствует или имеет крайне медленную скорость развития. Из-за поражения черепно-мозговых нервов может сформироваться слепота, и появиться нистагм. На фоне хронической внутричерепной гипертензии происходит задержка нейродвигательного развития. Для оценки неврологического статуса рекомендуется проведение компьютерной томографии или МРТ. Эти исследования позволяют измерить размеры церебральных желудочков, в т.ч. в динамике. При легких формах мукополисахаридоза неврологические нарушения могут проявляться только головными болями, которые неоднократно повторяются, а также умеренно выраженными зрительными нарушениями.
При мукополисахаридозе поражается не только головной мозг, но и спинной. Обычно это связано с вывихами позвонков. Сдавление нижерасположенных структур может приводить к слабости в нижних конечностях и нарушению походки.
- 3. Поражения опорно-двигательного аппарата.
Для мукополисахаридоза типично искривление в переднезаднем направлении поясничного сегмента позвоночного столба, в тяжелых случаях возможно развитие горба. Искривление может случиться и в боковой проекции (формируется сколиоз). Подвывихи в суставах приводят к деформации конечностей – они могут искривляться в наружную или внутреннюю сторону.
Нередко развивается карпальный туннельный синдром. Это состояние характеризуется сдавлением нервов увеличенными в объеме связками. Проявляется карпальный синдром болями и онемением кисти, отеком тканей. Боль и дискомфортные ощущения в запястной области, слабость мышц руки и онемение – частые симптомы туннельного синдрома, которые наблюдаются преимущественно у взрослых пациентов.
- 4. Дыхательная система.
У пациентов с мукополисахаридозом достаточно часто формируется тяжелая недостаточность дыхания, что связано с ограниченными возможностями легких расправляться. Также характерна обструктивная остановка дыхания во сне, бронхиальная астма. На фоне поражения легочной ткани повышаются риски инфекционных осложнений. Способствуют частым простудам увеличение языка и миндалин, узость трахеи, а также чрезмерное отложение гликозаминогликанов в слизистой оболочке дыхательных путей.
При мукополисахаридозе происходят поражения всех органов и систем. Поэтому симптоматика очень разнообразна, но вместе с тем и неспецифична.
Причины мукополисахаридоза
Непосредственная причина мукополисахаридоза связана с мутациями генов.
Практически все нозологические формы мукополисахаридоза имеют аутосомно-рецессивный тип передачи. Исключением из этого правила является только синдром Хантера. Последний передается в связке с Х-хромосомой, которая может быть носителем мутантного гена. Хантеровский синдром является орфанной патологией, для которой характерны метаболические нарушения. В настоящее время в мире насчитывается около 2000 человек с данным синдромом.
Из-за того, что заболевание наследуется по Х-сцепленному типу, заболевают только мальчики (в мужском генотипе всего 1 Х-хромосома, поэтому если она имеет дефект, то он проявит себя клинически). Девочки могут быть только бессимптомными носителями (у них 2 Х-хромосомы, и функцию больной – замещает здоровая). Вместе с тем в литературе описаны единичные случаи, когда хантеровский синдром развивался у женщин. Скорее всего, это связано с впервые возникшей мутацией гена в заинтересованном локусе.
Накапливающиеся при синдроме Хантера метаболиты гликозаминогликанов являются мощным стимулятором для иммунной системы, а также проявляют токсические свойства. в таких условиях организм «пытается» отгородиться от «неприятностей», поэтому активируется иммунитет, и развивается воспалительная реакция. Наиболее выражено асептическое воспаление в коже, сухожильном компартменте, сосудах, клапанах сердца и респираторном тракте.
Получить консультацию
Если у Вас наблюдаются подобные симптомы, советуем записаться на прием к врачу. Своевременная консультация предупредит негативные последствия для вашего здоровья.
Узнать подробности о заболевании, цены на лечение и записаться на консультацию к специалисту Вы можете по телефону:
Источник https://clinpharm-journal.ru/articles/2018-3/mukopolisaharidozy-put-k-diagnozu/
Источник https://www.smclinic.ru/diseases/mukopolisakharidoz/
Источник